의료기기 지침 (Medical Devices Directive, 93/42/EEC)
의료 기기에는 세 가지 지침들이 있습니다
- 능동형 체내 삽입용 의료기기 (AIMD) 지침: 90/385/EEC
- 의료기기 지침 (MDD) - 93/42/EEC
- 체외 진단용 의료기기 지침 (IVD) - 98/79/EC
다른 두 가지 의료기기 지침 하의 규정들도 상당 부분 동일하지만,
다음 설명은 MDD 규정을 인용합니다.
MDD에서 '의료 기기'란 다음 목적들을 위해 사람에게 쓰이도록 제조된
어떤 기구, 장치, 기기, 물질 또는 다른 물품들로 정의됩니다.
- 질병의 진단, 예방, 관찰, 치료 또는 완화,
- 상해 또는 장애에 대한 진단, 관찰, 치료, 완화 또는 보상
- 해부학적 또는 생리학적 과정의 조사, 교체 또는 수정
- 난소의 수정 제어,
그리고 인체 내외에서 약물학적, 면역학적 또는 신진 대사적 수단을 통해 그 자체의 주요한 동작을 해내지는 않지만, 그러한 수단을 통해 그 기능에 도움을 줄 수 있는 것으로 정의됩니다.
'부속품'이란, 그것이 제조자에 의해서 분명하게 특정 기기와 함께 사용되도록 의도되지 않았을지라도, 특정 의료기기가 제조자가 의도한 용도에 맞게 사용되기 위해서 함께 사용되는 물품을 의미합니다.
의료 기기 지침에서 '제조자'란 의료기기가 특정인의 이름 하에, 직접 또는 제3자에 의해서든 상관없이, 시장에 출하되기 전에 해당 기기의 설계, 제조, 포장 그리고 라벨링을 책임지는 사람을 의미합니다.
모든 의료기기는 MDD 부속서 I 에 서술된 적용가능한 안전, 수행, 라벨링 상의 '필수요건들'을 충족시켜야 합니다. 안전 요구사항들은 환자들만을 대상으로 한정되어 있지 않고 의료기기 사용자들에게도 적용됩니다.
라벨이있을 수 있으며, 일반적으로 그 국가의 언어 (들)에서 각 회원 국가의 필요, 기호의 사용을 권장합니다.
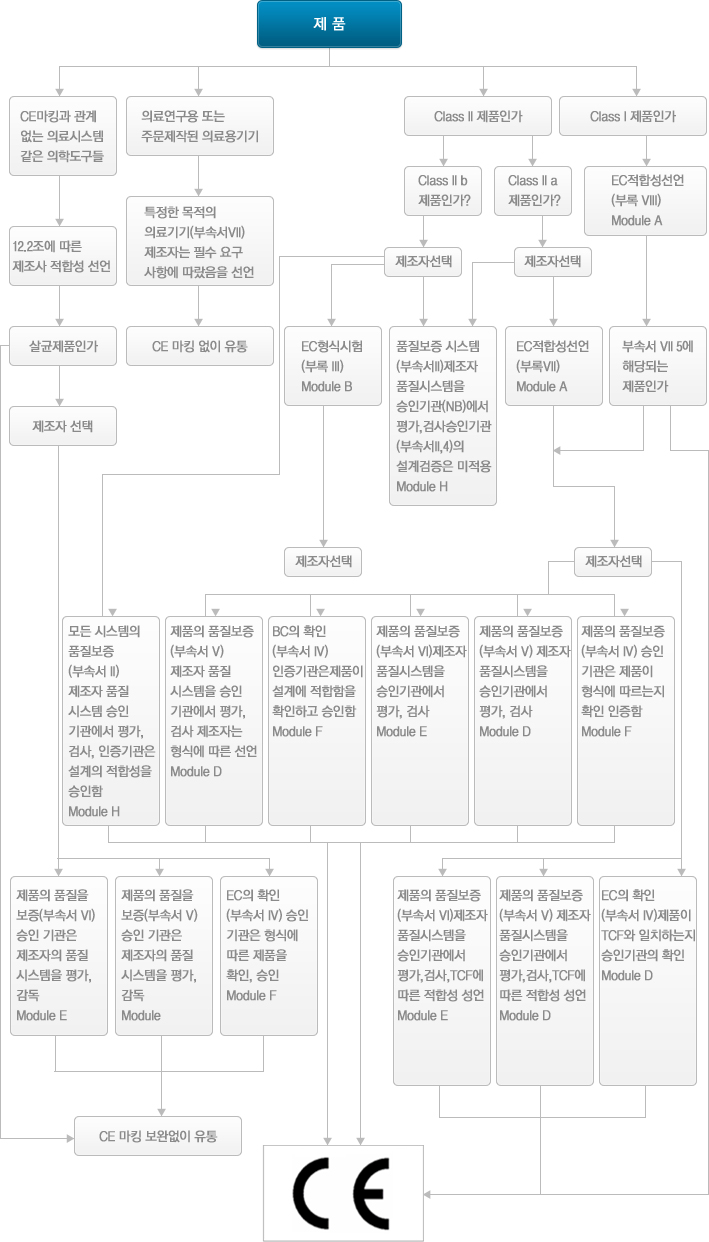
의료 기기는 MDD의 부속서 IX에 따라 분류됩니다. 분류 제조업체 MDD의 부속서 II, III, IV, V, VI와 VII에 따라 수행해야하는 적합성 평가 절차를 결정합니다.
의료기기 분류
MDD Article 9의 (1)에 따라 Ⅰ, Ⅱa, Ⅱb, Ⅲ의 네가지 등급으로 분류하며, 제조자가 분류 규칙(Annex 9)에 따라 자율적으로 분류. 만약, 분류에 대한 문제로 분쟁이 생길 경우 Article 9(2)에 따라 최종판단은 인증기관이 속한 국가의 법원에서의 판결에 의하는 것으로 규정되어 있다.
같은 품목이라도 사용목적과 효능, 사용되는 시간 그리고 구조나 원자재에 따라 등급은 달라질 수 있다.
※ 분류기준(Classification criteria)
- 기간(Duration) : 임시-60분 이내, 단기-30일 이하, 장기-30일 이상
- 삽입 의료기기(Invasive device) : 체강 또는 피부를 통하여 인체내에 전체 또는 부분적으로 삽입 되는 기기
- 작동 의료기기(Active device): 전기적인 에너지나 동력에 의해 사용되는 기기
※ 분류(Classification)
- 비삽입기기(Non-invasive devices) : Rule 1∼4
- 삽입기기(Invasive devices) : Rule 5∼8
- 작동기기(Active device) : Rule 9∼12
- 특별규칙(Special rules) : Rule 13∼18
임상보고서
일반적으로, 특징 및 공연 장치의 사용 정상적인 조건과 바람직하지 않은 부작용의 평가에 관한 요구 사항 적합성 확인은 '임상 데이터'를 기반으로해야합니다. 이 클래스 III에 implantable 장치 및 장치에 특히 적용됩니다. 별관 X에 따르면, 임상 데이터를 기반으로해야합니다.
- 장치의 의도 목적에 현재 사용할 수있는 관련 과학 문헌의 편집뿐만 아니라 고용 기술, 해당되는 경우,이 컴파일의 중요한 평가를 포함하는 서면 보고서 중
- 또는 모든 임상 조사의 결과는 실시하였다.
임상 조사 내용은 제 15 조 및 MDD 적용 및 임상 실험의 실적을 부속서 X에 누웠 규칙은 표준 EN 540을 따르도록 권장합니다.
다음은 통지 기관의 추천 NB-MED/2.7/Rec 1 ' 지도 와 NB-MED/2.7/Rec 3 '임상 데이터의 평가'clinicals에 '도움이 됩니다.
유럽대리인
MDD 제 EU 지역 클래스 I 또는 EU 시장에서 맞춤 제작 한 장치의 회원 국가에서 사업의 등록 장소가없는 제조업체, 그는 지역 사회에 설립 된 '책임자'를 지정한다, 그런 14.2 규칙 (예 권한을 부여받은 대리인). 이 사람은 사업의 등록 장소의 주소의 회원 국가와 관련 장치의 범주의 '관할 당국'을 통보하여야한다. 또한 라벨 또는 외부 포장, 또는 사용에 대한 지침은 또한에 책임을 사람 또는 커뮤니티 내에서 또는 지역 사회 내에서 설립 된 수입의 설립 제조업체의 권한을 부여받은 대리인 중 하나의 이름과 주소를 포함한다. 이 사람은 '기술 파일'의 저장에 관한 직무에 의무입니다
14 년부터 1998년 6월 MDD 93/42/EEC의 적용없이 의료 기기는 CE 마크를 운반하지 않는 시장에 위치한다. 이 새로운 또는 여부와 상관없이, 유통 및 / 또는 커뮤니티 시장에서 사용하는 방향으로 전망을 감상 할 수있는, 지불 또는 임상 조사를위한 장치가 아닌 다른 장치의 자유의 요금 대가로 사용할 수 있도록 수단 '시장에 배치' 완전히. 단장 CE 마크없이 장치 '를 서비스에 퍼팅'으로 2001년 6월 30 일까지 연장 전환 기간이있었습니다. 서비스로주는 장치가 의도 된 목적을 위해 처음으로 커뮤니티 시장에서 사용할 준비로서 최종 사용자에게 제공되었습니다되는 단계를 의미합니다.
CE 마크가 필요한 게 유일한 장치 제조업체가 MDD 부록 VIII에 따라 문서를 보관해야 '맞춤 제작 장치'와 '임상 조사를위한 장치'입니다. 주문 제작 장치는 구체적으로 자신의 책임을 특정 설계 특성에 따라, 제공하고 특정 환자의 단독 사용하기위한 것입니다 정당한 자격을 갖춘 의사의 서면 처방에 따라 작성된 모든 장치를 의미합니다.
의료기기지침(93/42/EEC)에 따른 적합성 절차
.jpg)





 About IEC KOREA
About IEC KOREA
.JPG)
.JPG)